Drug Repurposing using Artificial Intelligence
An unwelcome cyclone may have cancelled my hiking plans, but it gave me the opportunity to delve into the fascinating world of drug repurposing. This blog post explores how identifying new uses for existing drugs can dramatically accelerate and reduce the cost of bringing vital treatments to patients, and how recent advances in machine learning can further streamline this process.
Over the Easter long weekend, Tāmaki Makaurau (Auckland) was visited by Cyclone Tam, which meant my hiking trip was cancelled. At least it gave me some time to work on a few projects and finish this blog post I’d been meaning to do for a while!
Drug Repurposing
Traditional drug discovery is a marathon, often taking over a decade and requiring an investment of more than a billion dollars for each successful drug. This lengthy and costly process highlights the urgent need for more efficient strategies to combat disease. One such promising strategy is drug repurposing: identifying new therapeutic uses for existing drugs or those already undergoing clinical trials. By bypassing much of the early-stage development, and sometimes significant parts of clinical trials, drug repurposing offers the potential to significantly reduce both the time and financial cost associated with bringing new treatments to patients. Drug repurposing is ideal for addressing therapeutic needs in disease areas where financial incentives are limited (such as neglected tropical diseases and antibiotic development), or for rapid deployment in situations demanding swift treatment interventions for imminent public health emergencies, such as during the COVID-19 pandemic.
One well-known example of drug repurposing is sildenafil, originally developed to treat high blood pressure and angina (chest pain), and famously repurposed for erectile dysfunction after clinical trials revealed this unexpected side effect. Another is thalidomide, developed to treat morning sickness and found to cause birth defects, which is now used to treat cancers and skin disorders associated with leprosy. The crucial role of drug repurposing was further highlighted during the recent COVID-19 pandemic, where the corticosteroid dexamethasone emerged as a vital intervention in reducing mortality among severely ill patients.
Machine learning offers a novel method for drug repurposing. Using high-quality activity data for a target of interest, a machine learning model can be trained to predict active compounds based on their molecular structure. This trained model can then be used to screen libraries of compounds for potential repurposing.
The Drug Repurposing Hub
The Drug Repurposing Hub is a curated and annotated collection of drugs that have already received FDA approval or are currently undergoing clinical trials. Maintained by the Broad Institute, this resource serves as both a virtual library, providing comprehensive information on each compound, and a physical library, offering access to compound plates for experimental screening.
Antibiotic Discovery using Deep Learning
This post was inspired by the work of the Collins Lab at MIT, who used deep learning techniques to identify potential compounds for repurposing as treatments for antimicrobial-resistant bacteria. Their research addresses the escalating crisis of antibiotic resistance, a looming threat projected to cause 10 million deaths annually by 2050.
To tackle this challenge, the researchers assembled a primary training set of 2,335 molecules, experimentally screening them against E. coli BW25113 and identifying 120 compounds with antimicrobial activity. With the goal of creating a robust and generalizable model, they ensured the training set was structurally diverse. This data was then used to train a directed message-passing neural network (dMPNN) to create a classification model capable of predicting antimicrobial activity based on a molecule’s chemical structure. The trained model was subsequently used to make predictions on virtual libraries of compounds, including the Drug Repurposing Hub.
Ninety-nine molecules from the Drug Repurposing Hub that were predicted to be active were experimentally tested, with 51 displaying growth inhibition against E. coli. This process led to the identification of a particularly promising compound, the c-Jun N-terminal kinase inhibitor SU3327, which the researchers named halicin. Notably, halicin possesses a structure distinctly different from conventional antibiotics and demonstrated potent inhibitory activity against E. coli growth.
Chemprop
Central to this research is the open-source software package Chemprop. Developed primarily by researchers at MIT using the PyTorch framework, Chemprop harnesses the capabilities of message-passing neural networks (MPNNs) for molecular property prediction. Chemprop can be used both as a command-line interface (CLI) and a Python API. It implements a d-MPNN architecture, a type of graph neural network particularly effective for extracting relationships from molecular structures represented as graphs. An advantage of Chemprop is its ability to learn directly from molecular representations, such as SMILES strings converted into molecular graphs, eliminating the need for manual feature engineering, which can also introduce biases.
ChEMBL
Lacking access to a physical lab for experimental data, I needed an alternative approach for data gathering. For this, I used ChEMBL, a freely accessible and meticulously curated chemical database housing bioactivity data for drug-like molecules. It is maintained by the European Bioinformatics Institute (EBI), part of the European Molecular Biology Laboratory (EMBL) in the UK. Using the ChEMBL Webservice, I was able to specifically download data relevant to the target of interest. This retrieved data then served as the foundational dataset for training a machine learning model, which was subsequently employed to virtually screen the compounds within The Drug Repurposing Hub.
Imports
import subprocess
from pathlib import Path
import pandas as pd
from chembl_webresource_client.new_client import new_client
from rdkit import Chem
from rdkit.Chem import Draw
from tqdm import tqdm
Path("data").mkdir(exist_ok=True)
Path("models").mkdir(exist_ok=True)
Getting the Data from ChEMBL
Using the ChEMBL Webservice, I downloaded the data associated with a target for training a machine learning model using the three functions below. The first searches ChEMBL with a target name, and returns the target ChEMBL ID of the most similar target. The second downloads the activity data associated that the target ChEMBL ID, along with the molecular ChEMBL IDs. The third returns the canonical SMILES for the molecular ChEMBL IDs.
target = new_client.target
activity = new_client.activity
molecule = new_client.molecule
def get_target_chembl_id(target_name: str) -> str:
"""Get the ChEMBL ID of a target given its name."""
print(f'Searching for target: "{target_name}"')
target_query = target.search(target_name)
name, chembl_id = target_query[0]["pref_name"], target_query[0]["target_chembl_id"]
print(f"Target name: {name}, ChEMBL ID: {chembl_id}")
return chembl_id
def get_target_activity(chembl_id: str) -> pd.DataFrame:
"""Get the activity data for a target given its ChEMBL ID."""
print(f"Getting activity data for {chembl_id}")
activities = activity.filter(
target_chembl_id=chembl_id,
standard_type="IC50",
relation="=",
standard_units="nM",
).only(
"molecule_chembl_id",
"standard_value",
)
print(f"Found {len(activities)} activities")
activities_df = pd.DataFrame(
list(tqdm(activities[:100], desc="Processing Activities"))
)[["molecule_chembl_id", "standard_value"]]
activities_df["standard_value"] = pd.to_numeric(activities_df["standard_value"])
return activities_df
def get_molecule_data(molecule_chembl_ids: list[str]) -> pd.DataFrame:
"""Get the molecule data for a list of ChEMBL IDs."""
print(f"Getting molecule data for {len(molecule_chembl_ids)} ChEMBL IDs")
compounds_provider = molecule.filter(
molecule_chembl_id__in=molecule_chembl_ids
).only("molecule_chembl_id", "molecule_structures")
compounds_df = pd.json_normalize(
list(tqdm(compounds_provider, desc="Processing Compounds"))
)[["molecule_chembl_id", "molecule_structures.canonical_smiles"]].rename(
columns={"molecule_structures.canonical_smiles": "canonical_smiles"}
)
return compounds_df
Format the Data for Chemprop
Chemprop takes the data in the form of a table containing one column of SMILES strings, and one columns of activities. For this example I am creating a classification model, so the activity column is a binary classification (1 or 0). The first function takes the activity data and molecule data and merges them on the molecule ChEMBL ID, before cleaning the data up and creating the binary variable based on a cutoff. This cutoff can be changed depending on the target and dataset. I’ve always found the CLI usage of Chemprop easier, so the data is saved as a CSV file for training.
def create_training_data(
activity_data: pd.DataFrame, molecule_data: pd.DataFrame, nm_cutoff: float
) -> pd.DataFrame:
"""Create the training data for the model."""
print("Creating training data")
training_data = (
(
pd.merge(
left=activity_data,
right=molecule_data,
on="molecule_chembl_id",
how="left",
)
.dropna(how="any")
.drop(columns="molecule_chembl_id")
)
.groupby("canonical_smiles", as_index=False)
.agg("mean")
)
training_data["activity"] = (training_data["standard_value"] < nm_cutoff).astype(
int
)
actives = training_data["activity"].sum()
inactives = len(training_data) - actives
percentage = 100 * actives / len(training_data)
print(f"Actives: {actives} ({percentage:.2f}%), Inactives: {inactives}")
return training_data.drop(columns="standard_value")
def save_training_data(
training_data: pd.DataFrame, target_name: str, output_dir: Path
) -> None:
"""Save the training data to a CSV file."""
print(f"Saving training data to {output_dir}")
output_dir.mkdir(parents=True, exist_ok=True)
training_data.to_csv(output_dir / f"{target_name}_training_data.csv", index=False)
print(f"Training data saved to {output_dir / f'{target_name}_training_data.csv'}")
Download the Drug Repurposing Hub dataset
This function downloaded the Drug Repurposing Hub dataset, tidies the data up and saves it for making predictions using Chemprop.
def canon_smiles(smiles: str) -> str | None:
"""Convert SMILES string to canonical SMILES."""
mol = Chem.MolFromSmiles(smiles)
if mol is None:
return None
return Chem.MolToSmiles(mol, canonical=True)
def download_drug_repurposing_hub() -> None:
"""Download the Drug Repurposing Hub dataset."""
if Path("data/drug_repurposing_hub.csv").exists():
print("Drug Repurposing Hub dataset already downloaded")
return None
url = "https://storage.googleapis.com/cdot-general-storage/repurposing_samples_20240610.txt"
drug_repurposing_df = pd.read_csv(
url,
sep="\t",
skiprows=9,
)
drug_repurposing_df["smiles"] = drug_repurposing_df["smiles"].apply(canon_smiles)
drug_repurposing_df.dropna(subset=["smiles"], inplace=True)
drug_repurposing_df.drop_duplicates(subset=["smiles"], keep="first", inplace=True)
drug_repurposing_df.to_csv("data/drug_repurposing_hub.csv", index=False)
print("Drug Repurposing Hub dataset downloaded and processed")
print(f"Number of unique SMILES: {len(drug_repurposing_df['smiles'].unique())}")
Make Predictions on the Drug Repurposing Hub Data
As I mentioned above, I find the CLI usage of Chemprop easier (especially since the move from v1 to v2), so here I’m using subprocess to run Chemprop. These two functions train a Chemprop model, and make predictions on the Drug Repurposing Hub data.
def train_chemprop_model(target_name: str, data_dir: str, output_dir: str) -> None:
"""Train a ChemProp model using the training data."""
print(f"Training ChemProp model for {target_name}")
chemprop_train_args = [
"chemprop",
"train",
"--data-path",
f"{data_dir}/{target_name.replace(" ", "_")}_training_data.csv",
"--task-type",
"classification",
"--save-dir",
f"{output_dir}/{target_name.replace(" ", "_")}",
"--split-type",
"scaffold_balanced",
]
subprocess.run(chemprop_train_args, check=True)
def predict_with_chemprop(target_name: str, preds_dir: str, model_dir: str) -> None:
"""Predict using the ChemProp model."""
print(f"Predicting with ChemProp model for {target_name}")
chemprop_predict_args = [
"chemprop",
"predict",
"--test-path",
"data/drug_repurposing_hub.csv",
"--model-paths",
f"{model_dir}/{target_name.replace(" ", "_")}",
"--smiles-columns",
"smiles",
"--preds-path",
f"{preds_dir}/{target_name.replace(" ", "_")}_predictions.csv",
]
subprocess.run(chemprop_predict_args, check=True)
Visualize the Top Predictions
Once we’ve scored the compounds, we want to take a look at the top scoring compounds.
def visualize_top_predictions(search: str) -> None:
"""Visualize the top predictions."""
predictions_df = (
pd.read_csv(f"data/{search.replace(" ", "_")}_predictions.csv")
.sort_values("activity", ascending=False)
.head(9)
)
return Draw.MolsToGridImage(
[Chem.MolFromSmiles(s) for s in predictions_df["smiles"]],
legends=[f"{a:.2f}" for a in predictions_df["activity"]],
molsPerRow=3,
)
Create a Drug Repurposing Pipeline
This function runs the whole process for a specified target search.
def run_repurposing_pipeline(search: str, nm_cutoff=25) -> None:
"""Run the drug repurposing pipeline."""
download_drug_repurposing_hub()
target_chembl_id = get_target_chembl_id(search)
activity_data = get_target_activity(target_chembl_id)
molecule_data = get_molecule_data(activity_data["molecule_chembl_id"].tolist())
training_data = create_training_data(
activity_data, molecule_data, nm_cutoff=nm_cutoff
)
save_training_data(training_data, search.replace(" ", "_"), Path("data"))
train_chemprop_model(target_name=search, data_dir="data", output_dir="models/")
predict_with_chemprop(target_name=search, preds_dir="data", model_dir="models/")
Run the Drug Repurposing Pipeline
Malaria
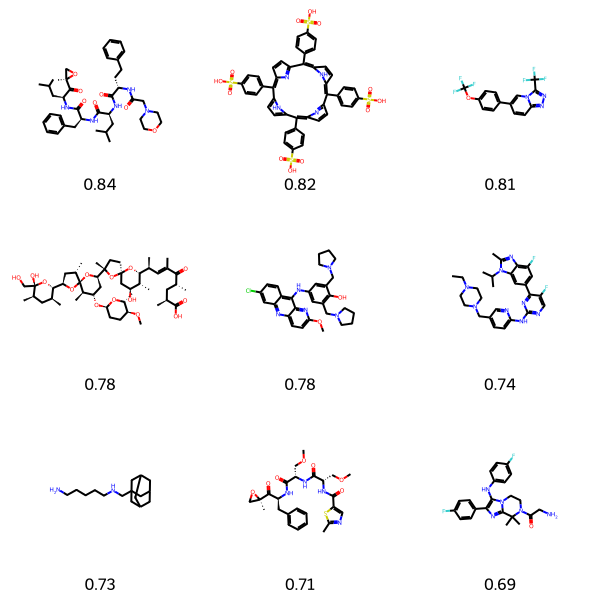
Malaria kills around half a million people per year, with 76% of global malaria deaths in children under 5 years old. It is considered a neglected tropical disease (NTD) due to its significant impact on global health, particularly in low-income countries. It disproportionately affects some of the world’s poorest people, and is often overlooked in global health efforts. Drug repurposing could offer alternative therapies significantly cheaper than developing an entirely new drug. Here I look to target the most deadly species of parasite that carries malaria - Plasmodium falciparum. After training the Chemprop model and screening the Drug Repurposing Hub, some compounds are flagged as potential hits and could be investigated experimentally.
Of the top 10 predictions, pyronaridine and KAF-156 are both malaria treatments, and were both in the training data, but interestingly pyronaridine was not classified as a hit in the training data. Carfilzomib, TPPS4, nanchangmycin, oprozomib, and lindane have all been investigated for treating malaria, so the model appears to be picking out the right compounds. Abemaciclib appears to have been recently investigated for repurposing with malaria. GS-967 appears to have no link to malaria yet, but maybe should be investigated experimentally!
search = "Plasmodium falciparum"
run_repurposing_pipeline(search)
visualize_top_predictions(search)
Drug Repurposing Hub dataset downloaded and processed
Number of unique SMILES: 6734
Searching for target: "Plasmodium falciparum"
Target name: Plasmodium falciparum, ChEMBL ID: CHEMBL364
Getting activity data for CHEMBL364
Found 45314 activities
Processing Activities: 100%|██████████| 45314/45314 [00:02<00:00, 15928.83it/s]
Getting molecule data for 45314 ChEMBL IDs
Processing Molecules: 100%|██████████| 21821/21821 [1:32:46<00:00, 3.92it/s]
Creating training data
Actives: 2677 (12.30%), Inactives: 19082
Saving training data to data
Training data saved to data/Plasmodium_falciparum_training_data.csv
Training ChemProp model for Plasmodium falciparum
Predicting with ChemProp model for Plasmodium falciparum
COVID-19
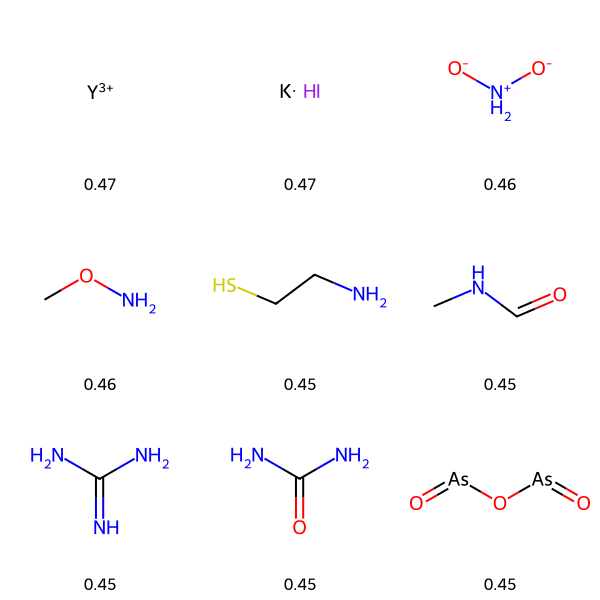
The COVID-19 pandemic has caused the deaths of over 7 million people so far, and in 2020 more than half of the world’s population was under some form of lockdown. With the speed that COVID spread around the globe, there was no time to develop new drug from scratch, and drug repurposing was explored in clinics to find treatments. In this example, I didn’t find any promising compounds in the Drug Repurposing Hub, but with a much smaller dataset than for malaria, the machine learning model is potentially much less powerfull.
search = "SARS-CoV-2"
run_repurposing_pipeline(search, nm_cutoff=500)
visualize_top_predictions(search)
Drug Repurposing Hub dataset already downloaded
Searching for target: "SARS-CoV-2"
Target name: SARS-CoV-2, ChEMBL ID: CHEMBL4303835
Getting activity data for CHEMBL4303835
Found 802 activities
Processing Activities: 100%|██████████| 802/802 [00:00<00:00, 12406.99it/s]
Getting molecule data for 802 ChEMBL IDs
Processing Compounds: 100%|██████████| 490/490 [00:00<00:00, 6081.22it/s]
Creating training data
Actives: 71 (14.55%), Inactives: 417
Saving training data to data
Training data saved to data/SARS-CoV-2_training_data.csv
Training ChemProp model for SARS-CoV-2
Predicting with ChemProp model for SARS-CoV-2